Text to go here...
Researchers have shown that they can treat sickle-cell anaemia in mice by switching on a haemoglobin gene usually only active before birth. This demonstrates that a technique called 'gene silencing' could potentially be used to treat sickle-cell anaemia in humans by targeting a protein that regulates the haemoglobin gene.
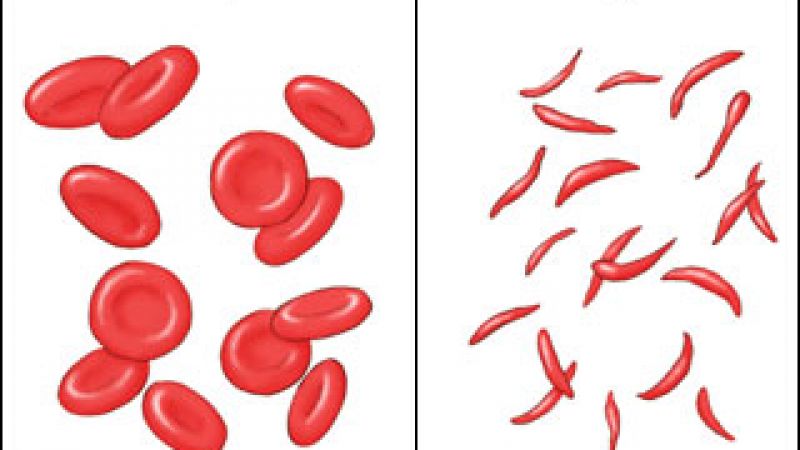
Sickle cell disease results from an abnormality in haemoglobin, the protein found in red blood cells responsible for transporting oxygen throughout the body. The defect causes the red blood cell to become stiff and misshapen. This can affect blood flow to tissues, damaging organs and causing pain. There are few treatments available.
The researchers were interested in a special form of haemoglobin, called fetal haemoglobin, which is only produced before birth. This form is not affected by sickle-cell disease. Previous research has found that a medicine called hydroxyurea raises levels of fetal haemoglobin after birth. This treatment reduces the pain and other symptoms associated with sickle-cell anaemia, but can have serious side effects and doesn't work in all patients. Doctors are therefore keen to find other ways of raising fetal haemoglobin levels.
Scientists discovered that a gene called BCL11A is involved in turning off the fetal haemoglobin gene shortly after birth. They believed that if they could switch off BCL11A they could keep the fetal haemoglobin gene turned on.
The scientists used a technique called 'gene silencing' to prevent protein being produced by the BCL11A gene in mice with sickle-cell anaemia. This allowed the fetal haemoglobin gene to remain active after birth preventing the mice suffering from the symptoms of sickle cell. Other aspects of blood production were unaffected.
Last edited: 29 July 2022 10:49